Maladie de Gaucher :
Vous n’êtes pas seul.
Les personnes atteintes de maladies rares vivent souvent avec des symptômes inexpliqués pendant des années*.
.
* La durée moyenne avant d’obtenir un diagnostic précis d’une maladie rare est de 4,8 ans.
Qu’est-ce que la maladie de Gaucher?
La maladie de Gaucher est un trouble génétique rare.
Elle est causée par une production insuffisante d’un type important de protéine appelée enzyme. Cette enzyme, appelée glucocérébrosidase, décompose une substance grasse appelée glucocérébroside. Lorsque le corps ne peut pas décomposer cette substance, des cellules appelées cellules de Gaucher, remplies de graisse, s’accumulent dans l’organisme.
La maladie de Gaucher est causée par une mutation d’un gène qui donne les instructions nécessaires à la fabrication d’une enzyme spécifique appelée glucocérébrosidase. Cette enzyme décompose normalement une substance grasse appelée glucocérébroside.
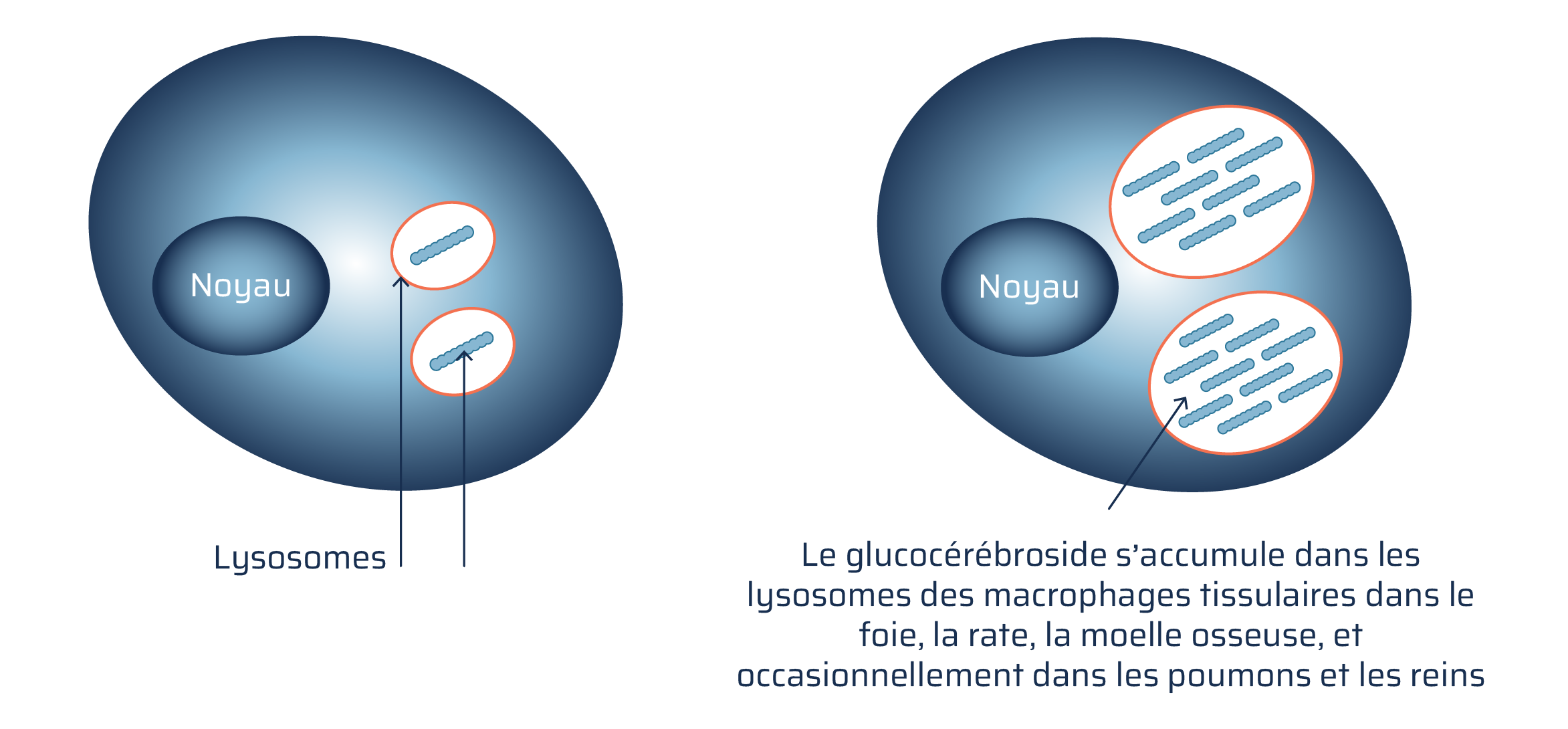
Une personne atteinte de la maladie de Gaucher n’a pas suffisamment de glucocérébrosidase; le glucocérébroside s’accumule donc dans les lysosomes, ce qui entraîne les symptômes associés à la maladie de Gaucher.
Macrophage normal contre cellule macrophage atteinte de la maladie de Gaucher
De 1 personne sur 40 000 à 1 personne sur 60 000 naît atteinte de la maladie de Gaucher
1 personne sur 800 dans la population juive ashkénaze
Signes et symptômes
La maladie de Gaucher est une maladie génétique rare qui touche de nombreux organes du corps. Les personnes peuvent présenter divers symptômes et être diagnostiquées à différents moments de leur vie, selon le type de maladie de Gaucher dont elles sont atteintes.
Les symptômes généraux comprennent :
- Douleur abdominale
- Douleur osseuse
- Fractures ou cassures des os
- Retard de croissance ou de puberté (chez les enfants)
- Ecchymoses faciles
- Ventre gonflé
- Perte d’appétit
- Faible nombre de globules rouges et de plaquettes
Le type 1 est la forme la plus courante de la maladie de Gaucher.
Quels signes et symptômes les personnes atteintes de la maladie de Gaucher de type 1 présentent-elles habituellement?
Cette illustration montre le pourcentage de personnes atteintes de la maladie de Gaucher de type 1 ayant déclaré chacun des symptômes :
![Une personne est représentée dans une illustration médicale avec ses organes internes et ses os visibles. Le foie et la rate sont mis en évidence, de même que les articulations du genou et de la hanche. Cette illustration montre le pourcentage de personnes atteintes de la maladie de Gaucher de type 1 ayant déclaré chacun des symptômes suivants : Douleur osseuse 63 % [pointe vers l’os du bras]; crise osseuse (diminution du flux sanguin vers le tissu osseux) 33 % [pointe vers l’os du bras]; hypertrophie de la rate 87 % [pointe vers les organes visibles dans la coupe]; hypertrophie du foie 79 % [pointe vers les organes visibles dans la coupe]; faible nombre de globules rouges 64 % [pointe vers les organes visibles dans la coupe]; faible numération plaquettaire 56 % [pointe vers le muscle de la jambe]; les os deviennent élargis et évasés (souvent près du genou) 46 % [pointe vers la radiographie du genou]; fracture due à l’affaiblissement causé par la maladie 15 % [pointe vers l’os du bras]; dégradation des articulations 8 % [pointe vers la radiographie de la hanche]; nécrose du tissu osseux 25 % [pointe vers la radiographie de la jambe]; faible densité minérale osseuse 42 % [pointe vers la radiographie de la jambe]; problèmes de moelle osseuse 40 % [pointe vers l’os de la jambe].](/dam/jcr:d41c6c2b-0c49-436c-a7b3-54cc7d6dc6dc/type1-gaucherdisease-FR-v1.2025-11-03-16-48-18.png)
Adapté et modifié d’après Charrow 2000, selon les données de personnes dans le registre de la maladie de Gaucher.
Vos symptômes pourraient-ils être liés à la maladie de Gaucher?
Trois types de maladie de Gaucher
Il existe trois types de maladie de Gaucher : type 1, type 2 et type 3.
Le type 1 représente 94 % de l’ensemble des cas de la maladie de Gaucher et ne touche habituellement pas le cerveau ni la moelle épinière, contrairement aux types 2 et 3.
Le type 2 est très rare; bien que les enfants puissent sembler normaux à la naissance, ils présentent des problèmes neurologiques vers l’âge de deux ans et leur état se détériore rapidement, entraînant souvent le décès durant la petite enfance.
Le type 3 présente souvent des caractéristiques similaires au type 1 ainsi qu’une progression plus lente que le type 2 et des patients atteignant généralement l’âge adulte.
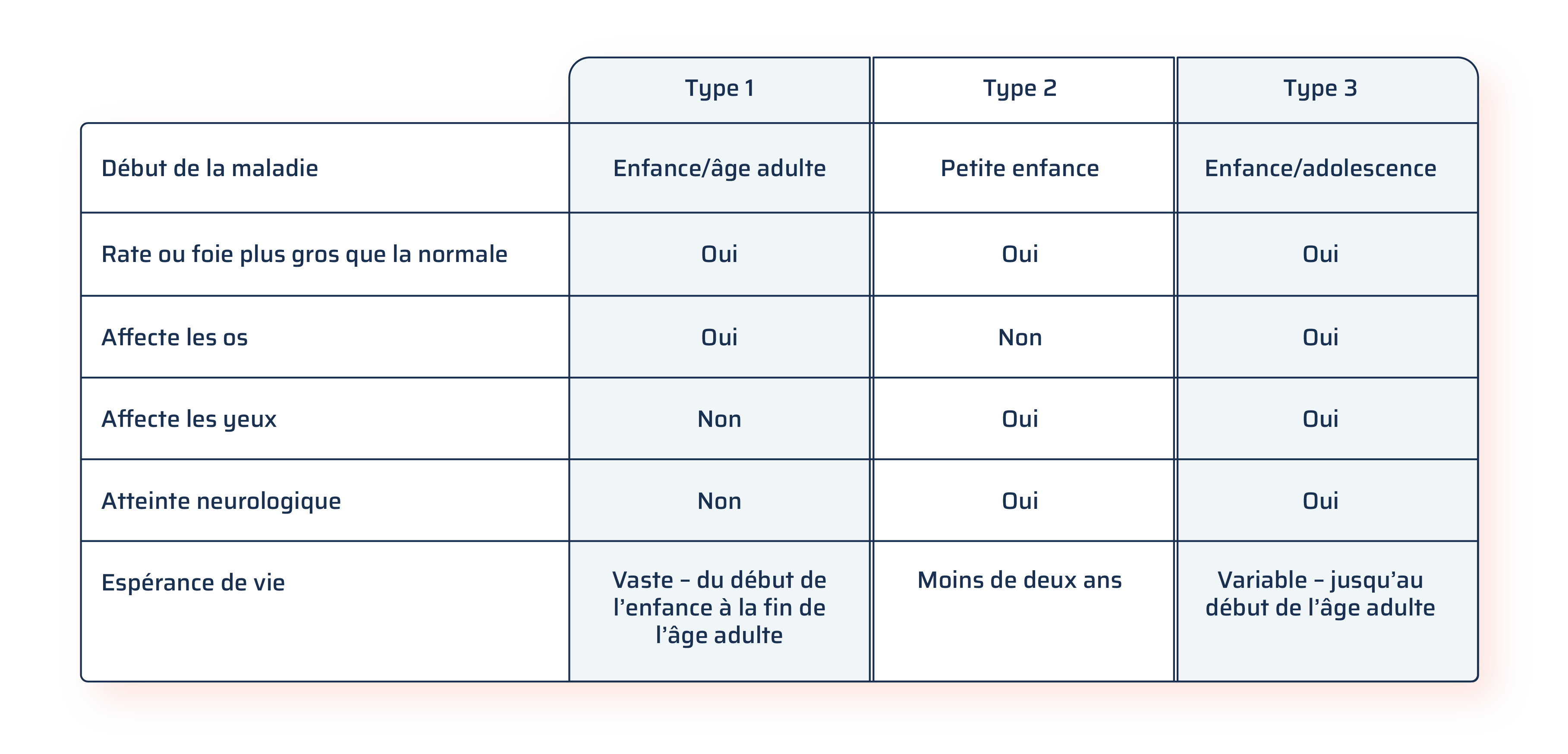
Adapté et modifié d’après Nagral 2014.
Comment la maladie de Gaucher est-elle diagnostiquée?
La maladie de Gaucher est d’origine génétique, ce qui signifie qu’elle est transmise par les parents. Cette maladie est ce qu’on appelle une affection autosomique récessive, ce qui signifie qu’une personne doit hériter de deux gènes atteints : un de sa mère et un de son père. Si vous héritez d’un seul gène, vous êtes porteur de la maladie et pourriez la transmettre à votre enfant, mais vous ne présenterez pas de symptômes.
Pour vérifier si vous êtes atteint de la maladie de Gaucher, votre médecin demandera une analyse de sang pour détecter l’enzyme glucocérébrosidase. Le laboratoire mesurera le niveau d’activité enzymatique dans l’échantillon de sang afin de confirmer ou d’écarter un diagnostic de maladie de Gaucher. Votre médecin pourrait aussi demander un test génétique pour confirmer le diagnostic.
Comment la maladie de Gaucher est transmise
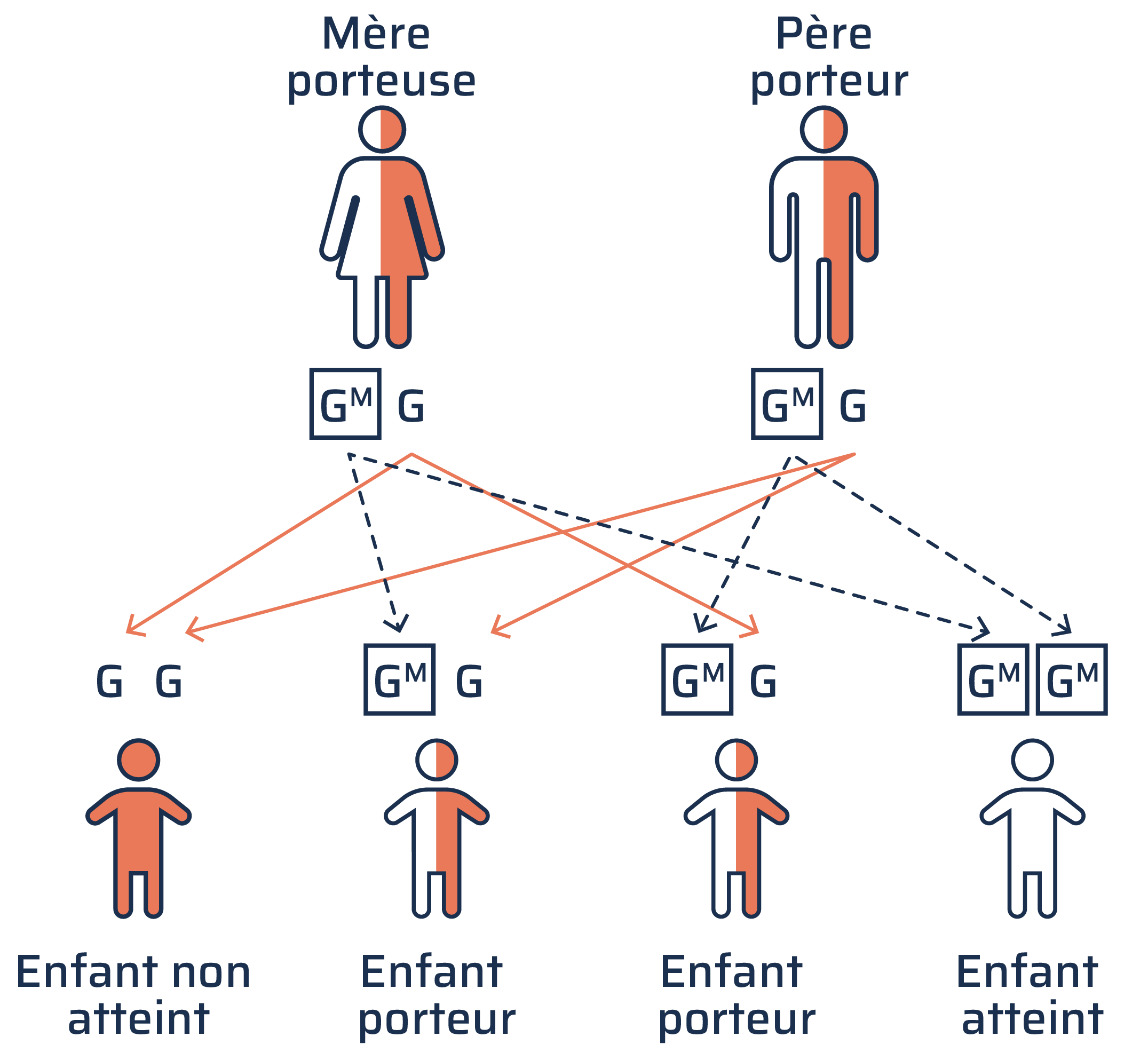
G = version normale du g.ne; production suffisante de b.ta-glucoc.r.brosidase pour d.grader le glucoc.r.broside. GM = version mut.e du g.ne; production insuffisante de b.ta-glucoc.r.brosidase pour d.grader le glucoc.r.broside.
Un diagnostic précoce est important, car il aide à réduire les dommages permanents au corps.
![]()
Environ 50 % des personnes atteintes de la maladie de Gaucher ont reçu leur diagnostic avant l’âge de 10 ans*
* 705/ 1441 personnes atteintes de la maladie de Gaucher inscrites au registre sur la maladie de Gaucher
Traitements
Il existe des traitements pour la maladie de Gaucher de type 1 et de type 3, mais il n’existe actuellement aucun traitement pour le type 2. Il existe deux principaux types de médicaments :
-
le traitement enzymatique substitutif (TES), qui fournit à l’organisme une enzyme pour réduire le glucocérébroside; et
-
le traitement par réduction du substrat (TRS), qui freine l’activité d’une enzyme participant à la production de glucocérébroside.
Ces traitements pourraient ne pas vous convenir et comportent un risque d’effets secondaires. Parlez à votre médecin.
